
.jpg "Nữ bệnh nhân hồi sinh nhờ trái tim người chết não")
.jpg)

.jpg)











.jpg "Huế: Cháy rừng dữ dội, gần 400 người tham gia dập lửa")

Gửi bình luận
Chủ Nhật, 20/7/2025
Chủ Nhật, 20/7/2025
Một bệnh nhân nữ 17 tuổi, nhập viện trong tình trạng thiếu máu rất nặng, xuất huyết toàn thân và khó thở được các bác sĩ chẩn đoán mắc hội chứng Evans, bệnh lý về máu hiếm gặp.
Chiều 6/6, Bệnh viện Quân y 175 cho biết, Khoa Huyết học lâm sàng và Bệnh nghề nghiệp của bệnh viện vừa điều trị thành công một ca bệnh hiếm gặp.
Bệnh nhân tên N.T.T.H, ngụ huyện Củ Chi, TP.HCM, có tiền sử gia đình với mẹ mắc giảm tiểu cầu miễn dịch.
3 năm gần đây, bệnh nhân thường xuyên xuất hiện các triệu chứng hoa mắt, chóng mặt, rong kinh kéo dài. Dù đã thăm khám tại nhiều cơ sở y tế và được chẩn đoán thiếu máu thiếu sắt, giảm tiểu cầu miễn dịch, điều trị bằng thuốc uống sắt nhưng không cải thiện.
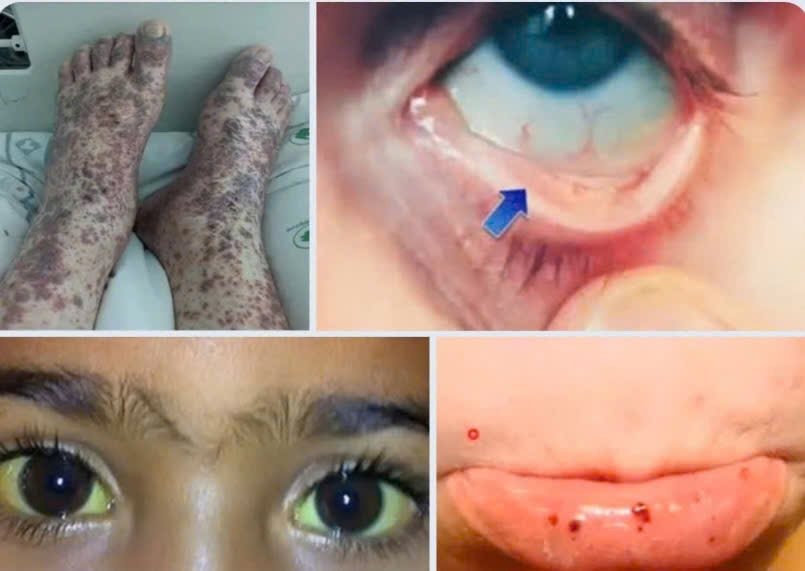
Một tuần trước nhập viện, bệnh nhân xuất hiện nốt xuất huyết vùng đùi, cẳng chân hai bên, lan rộng toàn thân kèm đau tức ngực, khó thở.
Bệnh nhân được đưa đến cấp cứu tại Bệnh viện Quân y 175 trong tình trạng thiếu máu nhược sắc mức độ rất nặng, với huyết sắc tố chỉ 31g/L, tiểu cầu giảm 8g/l kèm theo hội chứng xuất huyết và tan máu.
Sau khi thực hiện các xét nghiệm chuyên sâu, các bác sĩ xác định bệnh nhân mắc Hội chứng Evans nguyên phát, một rối loạn tự miễn hiếm gặp khiến cơ thể tự sinh kháng thể tấn công các tế bào máu như hồng cầu, tiểu cầu và đôi khi là bạch cầu.
Phác đồ điều trị được triển khai khẩn trương với truyền máu và phối hợp thuốc ức chế miễn dịch. Sau 11 ngày điều trị tích cực, các chỉ số huyết học cải thiện rõ rệt, huyết sắc tố tăng lên 1g/L, tiểu cầu đạt 177G/L.
Bệnh nhân xuất viện, tiếp tục theo dõi ngoại trú và tái khám định kỳ với sức khỏe ổn định.
Bác sĩ Mai Thị Thu Ly, Khoa Huyết học lâm sàng và Bệnh nghề nghiệp, Bệnh viện Quân y 175, người trực tiếp điều trị cho biết, hội chứng Evans là một trong những bệnh lý huyết học tự miễn hiếm gặp và phức tạp, chỉ chiếm khoảng 7% các trường hợp tan máu tự miễn và 2% các trường hợp giảm tiểu cầu miễn dịch.

Bệnh có biểu hiện lâm sàng rầm rộ, diễn tiến nhanh và dễ gây biến chứng nghiêm trọng nếu không được phát hiện sớm. Do triệu chứng không điển hình, nhiều trường hợp bị chẩn đoán nhầm với thiếu máu thông thường, bệnh lý gan, rối loạn đông máu hoặc các bệnh hệ thống khác, khiến xử trí chậm trễ.
Việc điều trị hội chứng Evans hiện nay còn nhiều thách thức do bệnh lý hiếm, thiếu các nghiên cứu đối chứng quy mô. Các phác đồ chủ yếu được suy rộng từ điều trị riêng lẻ của tan máu tự miễn và giảm tiểu cầu miễn dịch.
Do đó, việc cá thể hóa phác đồ điều trị, đánh giá toàn diện và phối hợp chặt chẽ giữa lâm sàng, xét nghiệm đóng vai trò quyết định trong kiểm soát bệnh.
Việc điều trị thành công bệnh nhân 17 tuổi tại Bệnh viện Quân y 175 là minh chứng rõ nét cho năng lực chuyên môn vững vàng, khả năng làm chủ các ca bệnh phức tạp của đội ngũ y bác sĩ.
Thời gian tới, bệnh viện sẽ tiếp tục đẩy mạnh phát triển chuyên khoa sâu, nâng cao chất lượng điều trị và khẳng định vị thế trung tâm y học quân sự đa năng, hiện đại, phục vụ hiệu quả sự nghiệp chăm sóc sức khỏe bộ đội và nhân dân.
Hội chứng Evans là một rối loạn tự miễn hiếm gặp, xảy ra khi cơ thể sinh kháng thể tấn công đồng thời hồng cầu (gây tan máu), tiểu cầu (gây xuất huyết) và đôi khi cả bạch cầu (gây suy giảm miễn dịch). Bệnh có thể xuất hiện riêng lẻ hoặc liên quan đến lupus ban đỏ hệ thống, hội chứng Sjogren, tăng sinh lympho...
Triệu chứng thường gặp bao gồm: da xanh, mệt mỏi, vàng da, xuất huyết dưới da, chảy máu chân răng, sốt, nhiễm trùng kéo dài. Gan và lách có thể to. Chẩn đoán dựa trên lâm sàng kết hợp xét nghiệm: Coombs dương tính, bilirubin gián tiếp tăng, haptoglobin giảm, LDH tăng, giảm hemoglobin và tiểu cầu.
Điều trị khởi đầu bằng corticosteroid, globulin miễn dịch tiêm tĩnh mạch. Trường hợp kháng trị có thể dùng rituximab, cyclosporin A hoặc phẫu thuật cắt lách. Điều trị hỗ trợ như truyền máu, truyền tiểu cầu cần được thực hiện thận trọng, theo dõi sát để phòng biến chứng miễn dịch.


.jpg)



CƠ QUAN NGÔN LUẬN CỦA TOÀ ÁN NHÂN DÂN TỐI CAO
Giấy phép số 226/GP-BTTTT do Bộ Thông tin và Truyền thông cấp
Tổng Biên tập: Trần Đức Vinh
Phó Tổng Biên tập: Tô Thị Lan Phương